Calculating Number of Iterations Required to Reach Steady-State
Selcen Ari
2020-11-26
Source:vignettes/convenient_iteration.Rmd
convenient_iteration.Rmd1. Introduction
In the ceRNAnetsim package, regulations of miRNA:target pairs are observed via direct or indirect interactions of elements in network. In this approach, change in expression level of single gene or miRNA can affect the whole network via “ripple effect”. So, when the change is applied the system, it affects to primary neighborhood firstly, and then propagates to further neighborhoods.
In the simple interaction network like minsamp, the ripple effect could be observed when expression level of Gene4 changes and subsequently effecting other genes. In the non-complex networks like minsamp, the steady-state condition can be provided easily, after network disturbed.
In this vignette, first, we demonstrate a suggestion to determine simulation iteration of existing dataset for gaining steady state after perturbing the network. Additionally, new approach which is useful for defining significant of nodes in terms of perturbation of network is elucidated.
2. Installation
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("ceRNAnetsim")
3. Comparison of gaining steady-state durations of middle and minimal datasets
data("minsamp")
minsamp %>%
priming_graph(competing_count = Competing_expression,
miRNA_count = miRNA_expression) %>%
update_how("Gene4",2) %>%
simulate_vis(title = "Minsamp: Common element as trigger", cycle = 15)
#> # A tbl_graph: 8 nodes and 7 edges
#> #
#> # A rooted tree
#> #
#> # Node Data: 8 x 7 (active)
#> name type node_id initial_count count_pre count_current changes_variable
#> <chr> <chr> <int> <dbl> <dbl> <dbl> <chr>
#> 1 Gene1 Competing 1 10000 10061. 10061. Competing
#> 2 Gene2 Competing 2 10000 10061. 10061. Competing
#> 3 Gene3 Competing 3 5000 5030. 5030. Competing
#> 4 Gene4 Competing 4 10000 19528. 19528. Competing
#> 5 Gene5 Competing 5 5000 5107. 5107. Competing
#> 6 Gene6 Competing 6 10000 10214. 10214. Competing
#> # … with 2 more rows
#> #
#> # Edge Data: 7 x 20
#> from to Competing_name miRNA_name Competing_expre… miRNA_expression dummy
#> <int> <int> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 1 7 Gene1 Mir1 10000 1000 1
#> 2 2 7 Gene2 Mir1 10000 1000 1
#> 3 3 7 Gene3 Mir1 5000 1000 1
#> # … with 4 more rows, and 13 more variables: afff_factor <dbl>,
#> # degg_factor <dbl>, comp_count_list <list>, comp_count_pre <dbl>,
#> # comp_count_current <dbl>, mirna_count_list <list>, mirna_count_pre <dbl>,
#> # mirna_count_current <dbl>, mirna_count_per_dep <dbl>, effect_current <dbl>,
#> # effect_pre <dbl>, effect_list <list>, mirna_count_per_comp <dbl>
minsamp %>%
priming_graph(competing_count = Competing_expression,
miRNA_count = miRNA_expression) %>%
update_how("Gene4",2) %>%
simulate(cycle = 5)
#> # A tbl_graph: 8 nodes and 7 edges
#> #
#> # A rooted tree
#> #
#> # Node Data: 8 x 7 (active)
#> name type node_id initial_count count_pre count_current changes_variable
#> <chr> <chr> <int> <dbl> <dbl> <dbl> <chr>
#> 1 Gene1 Competing 1 10000 10061. 10061. Up
#> 2 Gene2 Competing 2 10000 10061. 10061. Up
#> 3 Gene3 Competing 3 5000 5030. 5030. Up
#> 4 Gene4 Competing 4 10000 19528. 19528. Down
#> 5 Gene5 Competing 5 5000 5107. 5107. Up
#> 6 Gene6 Competing 6 10000 10214. 10214. Up
#> # … with 2 more rows
#> #
#> # Edge Data: 7 x 20
#> from to Competing_name miRNA_name Competing_expre… miRNA_expression dummy
#> <int> <int> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 1 7 Gene1 Mir1 10000 1000 1
#> 2 2 7 Gene2 Mir1 10000 1000 1
#> 3 3 7 Gene3 Mir1 5000 1000 1
#> # … with 4 more rows, and 13 more variables: afff_factor <dbl>,
#> # degg_factor <dbl>, comp_count_list <list>, comp_count_pre <dbl>,
#> # comp_count_current <dbl>, mirna_count_list <list>, mirna_count_pre <dbl>,
#> # mirna_count_current <dbl>, mirna_count_per_dep <dbl>, effect_current <dbl>,
#> # effect_pre <dbl>, effect_list <list>, mirna_count_per_comp <dbl>
Minsamp common target perturbation
For example, in minsamp dataset, the steady-state is occurred at iteration-14 (as seen above: after iteration-13, it is observed that there are only orange (miRNAs) and green (competing genes) nodes in network. In this case, genes have new regulated (steady) expression values while expression values of microRNAs are same in comparison with initial case.).
However, when network is larger and interactions are more complex, the number of iterations required to reach steady-state may increase. While at cycle 14 minsamp dataset has reached steady-state, the midsamp (middle sized sample) dataset has not reached steady-state after 15 cycles. In the example below, in midsamp data, Gene17 is upregulated 2 fold as a trigger and simulation is run for 15 cycles.
data("midsamp")
midsamp
#> Genes miRNAs Gene_expression miRNA_expression seeds targeting_region Energy
#> 1 Gene1 Mir1 10000 1000 0.43 0.30 -20
#> 2 Gene2 Mir1 10000 1000 0.43 0.01 -15
#> 3 Gene3 Mir1 5000 1000 0.32 0.40 -14
#> 4 Gene4 Mir1 10000 1000 0.23 0.50 -10
#> 5 Gene4 Mir2 10000 2000 0.35 0.90 -12
#> 6 Gene5 Mir2 5000 2000 0.05 0.40 -11
#> 7 Gene6 Mir2 10000 2000 0.01 0.80 -25
#> 8 Gene4 Mir3 10000 3000 0.43 0.40 -6
#> 9 Gene6 Mir3 10000 3000 0.35 0.90 -15
#> 10 Gene7 Mir3 7000 3000 0.23 0.30 -20
#> 11 Gene8 Mir3 3000 3000 0.01 0.20 -30
#> 12 Gene6 Mir4 10000 5000 0.05 0.40 -12
#> 13 Gene9 Mir4 6000 5000 0.32 0.80 -18
#> 14 Gene10 Mir4 2000 5000 0.43 0.20 -23
#> 15 Gene11 Mir4 8000 5000 0.35 0.90 -25
#> 16 Gene12 Mir4 1500 5000 0.43 0.40 -30
#> 17 Gene13 Mir4 500 5000 0.23 0.90 -17
#> 18 Gene14 Mir4 7000 5000 0.43 0.80 -15
#> 19 Gene14 Mir3 7000 3000 0.43 0.90 -25
#> 20 Gene15 Mir3 3000 3000 0.35 0.20 -12
#> 21 Gene16 Mir3 2000 3000 0.01 0.80 -18
#> 22 Gene17 Mir3 6000 3000 0.23 0.40 -22
#> 23 Gene17 Mir2 6000 2000 0.35 0.90 -7
#> 24 Gene18 Mir2 1000 2000 0.01 0.01 -30
#> 25 Gene19 Mir2 6500 2000 0.43 0.90 -32
#> 26 Gene20 Mir2 4800 2000 0.35 0.80 -18
midsamp %>%
priming_graph(Gene_expression, miRNA_expression) %>%
update_how("Gene17",2) %>%
simulate_vis(title = "Midsamp: Gene with higher degree as trigger", 15)
#> # A tbl_graph: 24 nodes and 26 edges
#> #
#> # A directed acyclic simple graph with 1 component
#> #
#> # Node Data: 24 x 7 (active)
#> name type node_id initial_count count_pre count_current changes_variable
#> <chr> <chr> <int> <dbl> <dbl> <dbl> <chr>
#> 1 Gene1 Competing 1 10000 10001. 10001. Up
#> 2 Gene2 Competing 2 10000 10001. 10001. Up
#> 3 Gene3 Competing 3 5000 5000. 5000. Up
#> 4 Gene4 Competing 4 10000 10110. 10110. Down
#> 5 Gene5 Competing 5 5000 5026. 5026. Down
#> 6 Gene6 Competing 6 10000 10105. 10105. Up
#> # … with 18 more rows
#> #
#> # Edge Data: 26 x 20
#> from to Competing_name miRNA_name Gene_expression miRNA_expression dummy
#> <int> <int> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 1 21 Gene1 Mir1 10000 1000 1
#> 2 2 21 Gene2 Mir1 10000 1000 1
#> 3 3 21 Gene3 Mir1 5000 1000 1
#> # … with 23 more rows, and 13 more variables: afff_factor <dbl>,
#> # degg_factor <dbl>, comp_count_list <list>, comp_count_pre <dbl>,
#> # comp_count_current <dbl>, mirna_count_list <list>, mirna_count_pre <dbl>,
#> # mirna_count_current <dbl>, mirna_count_per_dep <dbl>, effect_current <dbl>,
#> # effect_pre <dbl>, effect_list <list>, mirna_count_per_comp <dbl>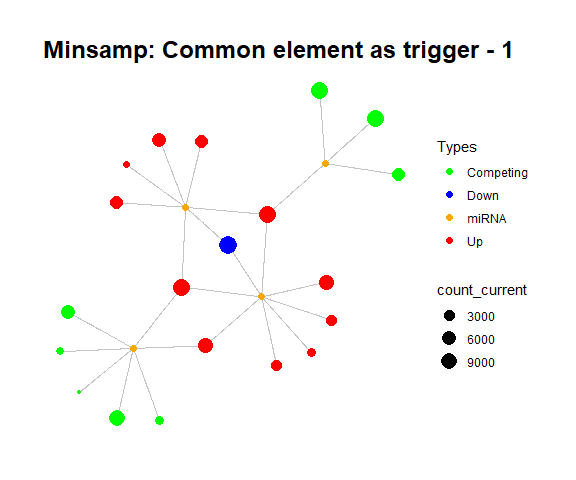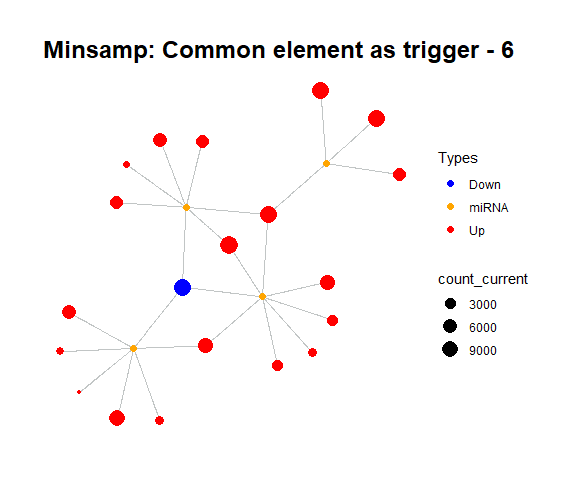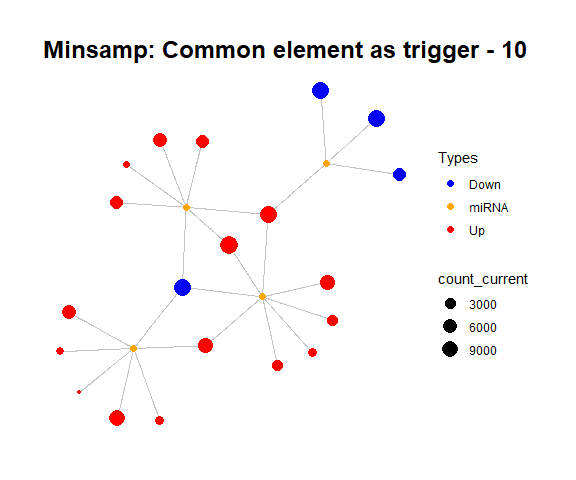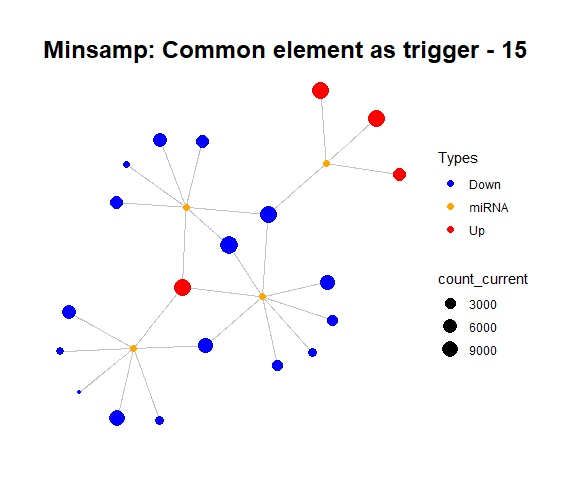
Midsamp Gene17 perturbation followed with 15 iterations
3.1. Suggestion for simulation iteration
Guessing or performing trial and error for large networks is not practical, thus we developed a function which calculates optimal iteration in a network after trigger and simulation steps. find_iteration() function analyses the simulated graph and suggests the iteration at which maximum number of nodes are affected. An important argument is limit which sets the threshold below which is considered “no change”, in other words, any node should have level of change greater than the threshold to be considered “changed”. Please be aware that small threshold values will cause excessively long calculation time especially in large networks.
In the example below, Gene2 is upregulated 2-fold and then iteration number at which maximum number of nodes affected will be calculated. The search for iteration number will go up to 50. Also, since we are searching for maximal propagation, limit is set to zero.
midsamp %>%
priming_graph(Gene_expression, miRNA_expression) %>%
update_how("Gene2",2) %>%
simulate(50) %>%
find_iteration(limit=0)
#> Warning in priming_graph(., Gene_expression, miRNA_expression): First column is processed as competing and the second as miRNA.
#> [1] 3NOTE: You can edit the dataset manually. You can change Gene2 expression value as 20000 and save that as a new dataset (midsamp_new_counts).
You can use the dataset that includes new expression values of miRNAs and genes.
data("midsamp_new_counts")
midsamp %>%
priming_graph(Gene_expression, miRNA_expression) %>%
update_variables(current_counts = midsamp_new_counts) %>%
simulate(50) %>%
find_iteration(limit=0)
#> Warning in priming_graph(., Gene_expression, miRNA_expression): First column is processed as competing and the second as miRNA.
#> [1] 1find_iteration() function will return a single number: the iteration number at which maximum propagation is achieved. If plot=TRUE argument is used then the function will return a plot which calculates percent of perturbed nodes for each iteration number. The latter can be used for picking appropriate number of cycles for simulate() function.
midsamp %>%
priming_graph(Gene_expression, miRNA_expression) %>%
update_how("Gene17",2) %>%
simulate(50) %>%
find_iteration(limit=0)
#> Warning in priming_graph(., Gene_expression, miRNA_expression): First column is processed as competing and the second as miRNA.
#> [1] 2
3.2. Find appropriate iteration number with find_iteration and then simulate accordingly
As shown in plot above, if “Gene17” is upregulated 2-fold, the network will need around 22 iterations to reach the steady-state. Since we have an idea about appropriate iteration number, let’s use simulate() function and iterate for 25 cycles using same trigger (Gene17 2-fold):
midsamp %>%
priming_graph(Gene_expression, miRNA_expression) %>%
update_how("Gene17", 2) %>%
simulate_vis(title = "Midsamp: Gene17 2 fold increase, 25 cycles", 25)
#> # A tbl_graph: 24 nodes and 26 edges
#> #
#> # A directed acyclic simple graph with 1 component
#> #
#> # Node Data: 24 x 7 (active)
#> name type node_id initial_count count_pre count_current changes_variable
#> <chr> <chr> <int> <dbl> <dbl> <dbl> <chr>
#> 1 Gene1 Competing 1 10000 10001. 10001. Competing
#> 2 Gene2 Competing 2 10000 10001. 10001. Competing
#> 3 Gene3 Competing 3 5000 5000. 5000. Competing
#> 4 Gene4 Competing 4 10000 10110. 10110. Competing
#> 5 Gene5 Competing 5 5000 5026. 5026. Competing
#> 6 Gene6 Competing 6 10000 10105. 10105. Competing
#> # … with 18 more rows
#> #
#> # Edge Data: 26 x 20
#> from to Competing_name miRNA_name Gene_expression miRNA_expression dummy
#> <int> <int> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 1 21 Gene1 Mir1 10000 1000 1
#> 2 2 21 Gene2 Mir1 10000 1000 1
#> 3 3 21 Gene3 Mir1 5000 1000 1
#> # … with 23 more rows, and 13 more variables: afff_factor <dbl>,
#> # degg_factor <dbl>, comp_count_list <list>, comp_count_pre <dbl>,
#> # comp_count_current <dbl>, mirna_count_list <list>, mirna_count_pre <dbl>,
#> # mirna_count_current <dbl>, mirna_count_per_dep <dbl>, effect_current <dbl>,
#> # effect_pre <dbl>, effect_list <list>, mirna_count_per_comp <dbl>
Midsamp Gene17 perturbation with 25 iteration
Note: If you ignore decimal change in gene expression, threshold argument can be used. With this method, system reaches steady-state early.
midsamp %>%
priming_graph(Gene_expression, miRNA_expression) %>%
update_how("Gene17", 2) %>%
simulate_vis(title = "Midsamp: Gene17 2 fold increase, 6 cycles", 6, threshold = 1)
#> # A tbl_graph: 24 nodes and 26 edges
#> #
#> # A directed acyclic simple graph with 1 component
#> #
#> # Node Data: 24 x 7 (active)
#> name type node_id initial_count count_pre count_current changes_variable
#> <chr> <chr> <int> <dbl> <dbl> <dbl> <chr>
#> 1 Gene1 Competing 1 10000 10001. 10001. Competing
#> 2 Gene2 Competing 2 10000 10001. 10001. Competing
#> 3 Gene3 Competing 3 5000 5000. 5000. Competing
#> 4 Gene4 Competing 4 10000 10110. 10110. Competing
#> 5 Gene5 Competing 5 5000 5026. 5026. Competing
#> 6 Gene6 Competing 6 10000 10105. 10105. Competing
#> # … with 18 more rows
#> #
#> # Edge Data: 26 x 20
#> from to Competing_name miRNA_name Gene_expression miRNA_expression dummy
#> <int> <int> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 1 21 Gene1 Mir1 10000 1000 1
#> 2 2 21 Gene2 Mir1 10000 1000 1
#> 3 3 21 Gene3 Mir1 5000 1000 1
#> # … with 23 more rows, and 13 more variables: afff_factor <dbl>,
#> # degg_factor <dbl>, comp_count_list <list>, comp_count_pre <dbl>,
#> # comp_count_current <dbl>, mirna_count_list <list>, mirna_count_pre <dbl>,
#> # mirna_count_current <dbl>, mirna_count_per_dep <dbl>, effect_current <dbl>,
#> # effect_pre <dbl>, effect_list <list>, mirna_count_per_comp <dbl>
Midsamp Gene17 perturbation with 6 iteration with threshold
The workflow that is aforementioned in this vignette should be considered as suggestion. Because the cycle is a critical argument that is used with simulate() function and affects results of analysis. In light of this vignette and functions, the approach can be developed according to dataset.
4. What is perturbation efficiency?
The perturbation efficiency means that the disturbance and propagation efficiency of an element in the network. In a given network not all nodes have same or similar perturbation efficiency. Changes in some nodes might propagate to whole network and for some nodes the effect might be limited to small subgraph of the network. Not only topology but also miRNA:target interaction dynamics determine perturbation efficiency.
Expression level and type of trigger element plays a crucial role. The trigger element can be an miRNA or competing target. The perturbation efficiency is affected from ratio of miRNA amount to sum of expression levels of its targets. Also, amount of competing element among whole competing elements is important since it determines distribution of miRNA. For example, if trigger is an miRNA with expression level of 1500 and if sum of expression levels of its targets is 1000000, then this miRNA will not perturb its neighborhood efficiently. So, miRNA:target ratio is important for regulation of interaction network.
In a biological system, the miRNA:target interactions does not depend solely on stoichiometry, unfortunately. miRNAs affect the targets via degradation or inhibition after the binding. The experimental studies have shown that the features of miRNA:target interactions determine the binding and degradation efficiency. For example, binding energy between miRNA and target and seed structure of miRNA determine the binding efficiency of complex. In addition, the binding region on the target affects the degradation of target.
On the other hand, interaction factors affecting binding and degradation of miRNA to its targets also have impact one efficiency of perturbation of the change. For instance, if a node has very low binding affinity to targeting miRNA, change in expression level of that node will cause weak or no perturbation.
Thus, we developed functions which can calculate perturbation efficiency of a given node or all nodes. calc_perturbation() function calculates perturbation efficiency for given trigger (e.g. Gene17 2-fold). find_node_perturbation() function screens the whole network and calculate perturbation efficiency of all nodes.
4.1. How does the calc_perturbation() work?
This function works for a given node from network. It calculates and returns two values:
- perturbation efficiency : mean of percentage of expression changes of all elements except trigger in the network
- perturbed count : count of disturbed nodes for given iteration number and limit (the minimum change in expression level needed to be considered “disturbed” or “changed”).
In the example below, “Gene17” is up-regulated 3-fold in midsamp dataset where Energy and seeds columns are used for calculating affinity effect and targeting_region columns is used for calculating degradation effect. The network will be iterated over 30 times and number of disturbed nodes (as taking into account nodes that have changed more than the value of the threshold (0.1 percentage in terms of the change)) will be counted.
midsamp %>%
priming_graph(competing_count = Gene_expression,
miRNA_count = miRNA_expression,
aff_factor = c(Energy,seeds),
deg_factor = targeting_region) %>%
calc_perturbation("Gene17", 3, cycle = 30, limit = 0.1)
#> Warning in priming_graph(., competing_count = Gene_expression, miRNA_count = miRNA_expression, : First column is processed as competing and the second as miRNA.
#> # A tibble: 1 x 2
#> perturbation_efficiency perturbed_count
#> <dbl> <dbl>
#> 1 0.377 8If you are interested in testing various fold change values of a given node, then we can use map (actually parallelized version future_map) to run function for set of input values.
First, let’s keep the primed version of graph in an object
primed_mid <- midsamp %>%
priming_graph(competing_count = Gene_expression,
miRNA_count = miRNA_expression,
aff_factor = c(Energy,seeds),
deg_factor = targeting_region)
#> Warning in priming_graph(., competing_count = Gene_expression, miRNA_count = miRNA_expression, : First column is processed as competing and the second as miRNA.Now, let’s calculate perturbation efficiency caused by 2-fold to 10-fold increase in Gene17
# for parallel processing
# future::plan(multiprocess)
seq(2,10) %>%
rlang::set_names() %>%
furrr::future_map_dfr(~ primed_mid %>% calc_perturbation("Gene17", .x, cycle = 30, limit = 0.1), .id="fold_change")If you’re interested in screening nodes instead of fold changes then you don’t have to write a complicated map command, there’s already a function available for that purpose.
4.2. A Short-cut: Finding perturbation efficiencies for whole nodes of network
The find_node_perturbation() function calculates the perturbation efficiency and perturbed node count of each node in network.
In the example below, each node is increased 2-fold and tested for perturbation efficiency for 4 cycles with threshold of 0.1
midsamp %>%
priming_graph(competing_count = Gene_expression,
miRNA_count = miRNA_expression,
aff_factor = c(Energy,seeds),
deg_factor = targeting_region) %>%
find_node_perturbation(how = 3, cycle = 4, limit = 0.1)%>%
select(name, perturbation_efficiency, perturbed_count)
#> # A tibble: 24 x 3
#> name perturbation_efficiency perturbed_count
#> <chr> <dbl> <dbl>
#> 1 Gene1 0.0626 2
#> 2 Gene2 0.0974 3
#> 3 Gene3 0.0311 2
#> 4 Gene4 0.561 10
#> 5 Gene5 0.0366 3
#> 6 Gene6 0.505 12
#> 7 Gene7 0.251 6
#> 8 Gene8 0.00973 0
#> 9 Gene9 0.688 6
#> 10 Gene10 0.510 6
#> # … with 14 more rowsOn the other hand, some of nodes in network might not be affected from perturbation because of low expression or weak interaction factors. In this case, fast argument can be used. Argument fast calculate affected expression percent of the targets and perturbation calculation is not ran for these elements in network, if that percentage value is smaller than given fast value.
midsamp %>%
priming_graph(competing_count = Gene_expression,
miRNA_count = miRNA_expression,
aff_factor = c(Energy,seeds),
deg_factor = targeting_region) %>%
find_node_perturbation(how = 3, cycle = 4, limit = 0.1, fast=5)%>%
select(name, perturbation_efficiency, perturbed_count)
#> Warning in priming_graph(., competing_count = Gene_expression, miRNA_count = miRNA_expression, : First column is processed as competing and the second as miRNA.
#> Subsetting by edges
#> Warning: `cols` is now required when using unnest().
#> Please use `cols = c(eff_count)`
#> # A tibble: 24 x 3
#> name perturbation_efficiency perturbed_count
#> <chr> <dbl> <dbl>
#> 1 Gene1 NA NA
#> 2 Gene2 NA NA
#> 3 Gene3 NA NA
#> 4 Gene4 NA NA
#> 5 Gene5 NA NA
#> 6 Gene6 1.26 8
#> 7 Gene7 NA NA
#> 8 Gene8 NA NA
#> 9 Gene9 3.12 8
#> 10 Gene10 2.90 8
#> # … with 14 more rowsThe results of the find_node_perturbation() will list effectiveness or importance of nodes in the network. This function can help selecting nodes which will have effective perturbation in network.
5. Session Info
sessionInfo()
#> R version 4.0.2 (2020-06-22)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 16.04.6 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/openblas-base/libblas.so.3
#> LAPACK: /usr/lib/libopenblasp-r0.2.18.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] purrr_0.3.4 png_0.1-7 ceRNAnetsim_1.3.2 tidygraph_1.2.0
#> [5] dplyr_1.0.2
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.1.0 xfun_0.19 listenv_0.8.0 graphlayouts_0.7.1
#> [5] colorspace_2.0-0 vctrs_0.3.5 generics_0.1.0 htmltools_0.5.0
#> [9] viridisLite_0.3.0 yaml_2.2.1 utf8_1.1.4 rlang_0.4.9
#> [13] pkgdown_1.6.1 pillar_1.4.7 glue_1.4.2 tweenr_1.0.1
#> [17] lifecycle_0.2.0 stringr_1.4.0 munsell_0.5.0 gtable_0.3.0
#> [21] ragg_0.4.0 future_1.20.1 codetools_0.2-16 memoise_1.1.0
#> [25] evaluate_0.14 labeling_0.4.2 knitr_1.30 parallel_4.0.2
#> [29] fansi_0.4.1 furrr_0.2.1 Rcpp_1.0.5 scales_1.1.1
#> [33] desc_1.2.0 farver_2.0.3 parallelly_1.21.0 systemfonts_0.3.2
#> [37] fs_1.5.0 textshaping_0.2.1 gridExtra_2.3 ggforce_0.3.2
#> [41] ggplot2_3.3.2 digest_0.6.27 stringi_1.5.3 ggrepel_0.8.2
#> [45] polyclip_1.10-0 rprojroot_2.0.2 grid_4.0.2 cli_2.2.0
#> [49] tools_4.0.2 magrittr_2.0.1 tibble_3.0.4 ggraph_2.0.4
#> [53] crayon_1.3.4 tidyr_1.1.2 pkgconfig_2.0.3 ellipsis_0.3.1
#> [57] MASS_7.3-51.6 assertthat_0.2.1 rmarkdown_1.15 viridis_0.5.1
#> [61] R6_2.5.0 globals_0.14.0 igraph_1.2.6 compiler_4.0.2